What to Expect During Pulmonary Embolism Recovery - Healthline
What to Expect During Pulmonary Embolism Recovery - Healthline |
- What to Expect During Pulmonary Embolism Recovery - Healthline
- Pulmonary Arterial Hypertension: Updates in Epidemiology and Evaluation of Patients - AJMC.com Managed Markets Network
- Right-Sided Heart Failure: Symptoms, Causes, and Treatments - Healthline
- Right sided heart failure: Symptoms, outlook, treatment - Medical News Today
| What to Expect During Pulmonary Embolism Recovery - Healthline Posted: 19 May 2021 12:00 AM PDT  A pulmonary embolism (PE) is when a blood clot becomes stuck in the blood vessels of your lung. These clots typically begin in the leg and then break free and travel to the lung. The American Lung Association estimates that about 1 in 1,000 people in the United States experience a PE each year. A PE can be a serious or life threatening condition, which means receiving prompt medical treatment is vital. Treatment of a PE focuses on making sure that the current clot doesn't get any bigger while also preventing new clots from forming. Recovery from a PE can take several weeks or months. Continue reading to learn more about:
The exact amount of time that it takes to recover from a PE can vary from person to person. Many people can completely recover and return to their normal level of activity after a period of several weeks or months. It's possible that some of your symptoms will ease as you receive treatment and your body heals. However, it's not uncommon to continue to have shortness of breath or chest pain for weeks, months, or even years after a PE. A 2019 study looked at quality of life in 101 people who'd had a PE. It found that 6 months after a PE, 47 percent of participants reported lingering shortness of breath and 25.3 percent reported some type of impairment or difficulty in functioning. Next, we'll go over some of the important factors that can impact how long your recovery will take. Severity of your PEThe severity of a PE can affect treatment options. For example, someone with a severe or life threatening PE may require more intensive treatment with thrombolytic medications or a medical procedure. These can potentially prolong your recovery time. Your overall healthYour overall health is important in the treatment and recovery process of any health condition. This is also true with PE. Certain underlying health conditions may put you at an increased risk for experiencing prolonged shortness of breath or difficulty with physical exertion after a PE. Some examples of these include: Blood clot riskA big part of the recovery for PE aims to prevent additional blood clots from forming. There are several risk factors that can increase your risk for blood clots, such as: Generally speaking, the more risk factors you have, the higher your risk is for developing a blood clot. As you're recovering from a PE, your doctor will assess your risk for future blood clots. Those at a higher risk level may need to take blood-thinning medications for a longer period. Many people who have a PE spend some time in the hospital to receive treatment. The length of this stay can depend on the severity of the PE. One study from 2008 found that the median length of hospital stay for a PE was 6 days. In some cases, it may be possible to receive treatment at home. The American Society of Hematology published guidelines in 2020 suggesting that physicians offer home treatment for those with PE who are at a low risk for complications. Next, let's review some of the treatments and follow-up care that you may receive after a PE. MedicationsThe primary treatment for a PE is the use of blood-thinning medications, also known as anticoagulants. Blood-thinning medications work to stop existing clots from getting larger and also prevent new clots from forming. However, they don't dissolve clots. Your body typically does this on its own over time. These medications can be given in pill form or as an injection. An example of a common blood thinner is warfarin. Other, newer blood-thinning medications are also available. In situations when a PE has become life threatening, clot-busting medications called thrombolytics may be given prior to blood-thinning medications. These strong medications work to dissolve blood clots. At a minimum, you'll typically have to take a blood-thinning medication for 3 months. Some people, including those at a higher risk for another serious blood clot, may need to continue taking it for a longer period of time. Medical proceduresSometimes, a medical procedure may be needed as a part of PE treatment. These can include:
Follow-up visitsYou'll have regular follow-up appointments with your doctor during your recovery period. These can begin anywhere from 2 weeks to 3 months after your PE. During these appointments, your doctor will evaluate how your recovery is progressing. They'll also address any questions or concerns that you may have. Blood tests are used to help your doctor gauge how well your medications are working and how well your body is tolerating them. Additional imaging isn't typically necessary unless you're having persistent symptoms like shortness of breath or fatigue. Three months after your PE diagnosis, your doctor will consider whether you need to continue on with blood-thinning medications. If you're at a low risk for future blood clots, you may not need to continue taking them. Lifestyle tipsTo promote cardiovascular health and prevent another blood clot from forming, it's important to implement various lifestyle adjustments during your recovery period and beyond. These can include things like: After a PE, it's normal to have questions and concerns about when you can safely return to your normal activities. The short answer is it depends on your overall condition as well as your doctor's recommendation. Let's look at some general guidelines about returning to your normal activity levels. Day-to-day activitiesAfter a PE, it's important to try to go about your daily activities when possible. During this time, listen to what your body is telling you. If a certain activity leaves you feeling short of breath or in pain, stop doing it and rest until you feel better. Returning to workWhen you're able to return to work can largely depend on the type of job that you have. In some cases, it may be possible to return to work within weeks. Your doctor will work with you to decide when it's appropriate to start working again. ExercisePhysical activity is often encouraged after a PE, as it can improve both circulation and lung function. It may be a good idea to start out with low-intensity activities, such as walking or yoga. As you recover, you can slowly begin increasing the intensity of your activities. During your follow-up appointments, ask your doctor for exercise recommendations. Based on the progression of your recovery, they can help give you an idea of what level of physical activity is appropriate. It's generally best to avoid strenuous exercise after a PE. There are a couple of reasons for this. First, your body needs time to heal and recover. Second, blood-thinning medications can increase your risk for serious bruising or bleeding if you're injured. As mentioned earlier, it's important to listen to your body during this time. Try to be patient and don't push yourself too hard, too soon. TravelingFlying, especially long-haul flights, isn't recommended during the first 4 weeks of your recovery. After this period, it's typically fine to travel, but it may be a good idea to discuss your travel plans with your doctor ahead of time. Whether you're traveling by car or by plane, it's important to make sure that you don't sit for too long. Take some time every couple of hours to get up and walk around for a few minutes. Here are some potentially serious complications that are important to keep an eye out for as you recover from a PE. BleedingIncreased bleeding is a side effect of blood-thinning medications. This can be serious, so seek medical attention right away if you notice any of the following while on blood thinners: A repeat DVT or PEAccording to the National Heart, Lung, and Blood Institute, nearly 1 in 3 people who have a venous blood clot, like deep vein thrombosis (DVT) or PE, will experience another in the next 10 years. Because of this, it's important to look out for the symptoms of these conditions and seek prompt medical care if they occur. Some potential signs of DVT include the following symptoms: Symptoms that mean you may be experiencing another PE include: Pulmonary hypertensionIn some people who have had a PE, scar tissue can form in nearby arteries, causing them to become narrower. This can lead to a condition called pulmonary hypertension. The symptoms of pulmonary hypertension include: Pulmonary hypertension can lead to heart failure, so it's important that it's treated. The American Lung Association recommends making an appointment with your doctor to be tested for pulmonary hypertension if you still have trouble with breathing 6 months after your PE. The outlook for those who've had a PE is generally good if it's detected and treated quickly. If not, PE can become life threatening. In fact, with prompt care, mortality from PE drops from 30 percent to 8 percent. The recovery period can vary by individual. While many people can recover completely over a period of weeks or months, others may take longer. Factors that influence recovery time include:
Some ways to help improve your outlook as you recover from a PE include:
As you recover and increase your activity levels, be sure to pay attention to what your body is telling you. Your doctor will work with you to help determine when it's appropriate to do things like return to work, travel, or engage in more strenuous activities. |
| Posted: 11 Mar 2021 12:00 AM PST  Am J Manag Care. 2021;27(3):S35-S41. https://doi.org/10.37765/ajmc.2021.88609 Introduction Pulmonary hypertension (PH) describes a group of severe pulmonary vascular disorders characterized by elevated mean pulmonary arterial pressure (mPAP) at rest.1 The World Symposium on Pulmonary Hypertension (WSPH) categorizes pulmonary hypertension into 5 groups (Table 1).2 Pulmonary arterial hypertension (PAH), which corresponds to group 1 PH, and a focus of this article, is a complex and devastating disease that causes progressive vasoconstriction and vascular remodeling of the distal pulmonary arteries.3 Currently, there is no cure, and the majority of patients with PAH go on to develop right heart dysfunction leading to death. Due to the progressive nature of PAH, it is crucial that the disease is diagnosed early with an accurate classification. Patients with PAH also must undergo a thorough evaluation to ascertain the severity of disease and future risk, and ideally have access to treatment at specialized care centers.1 The past 2 decades have been marked by significant advancements leading to novel therapeutics and improved understanding of the pathogenesis of PAH. As a result, the management of PAH is rapidly evolving. Classification and Etiology of PAH PAH includes several subgroups, all having similar pulmonary vascular pathobiology, clinical characteristics, and management strategies (Table 2).4-6 PAH can be idiopathic, heritable, caused by drugs or toxins, or associated with other conditions such as connective tissue disease, congenital heart disease, or pulmonary hypertension.1 Idiopathic PAH is responsible for more than 50% of all PAH cases. It requires extensive investigation (diagnosis of exclusion), whereas heritable PAH results from gene mutations or familial cases regardless of mutations.7 The 6th WSPH updated the group 1 pulmonary hypertension classification to include new drugs and toxins as known agents associated with PAH.6 For example, amphetamines, methamphetamines, and dasatinib were added to the definite association category.8 Leflunomide, bosutinib, and direct-acting antivirals for hepatitis C virus (eg, sofosbuvir) were added as agents having a possible association.9-15 A small subset of patients with PAH presenting with overt features of venous/capillary involvement was also recognized as a distinct category. Pulmonary veno-occlusive disease and/or pulmonary capillary hemangiomatosis (PVOD/PCH) were moved to a subset within group 1 as opposed to the prior iteration of the WSPH, where they were designated as group 1.1,2 Typically, individuals with PVOD/PCH have similar clinical presentations and hemodynamic profiles as those with PAH; however, they have poorer prognosis, limited response to PAH treatment, and are at high risk for developing pulmonary edema from PAH therapeutics.16 A separate group of individuals with long-term response to calcium channel blockers was included in the updated clinical classification based on accumulating improved survival data in this small subset of patients.1,17 Epidemiology PAH is a rare disorder found in 15 to 50 persons per million within the United States and Europe.1 Idiopathic, heritable, and anorexigen-induced PAH make up 52.6% of all PAH cases. Generally, PAH affects women aged between 30 and 60 years. However, it can occur in males and is often associated with worse clinical outcomes.7 The National Institutes of Health (NIH) was a landmark registry that collected PAH data between 1981 and 1985.5,18 This registry included 187 individuals having PAH of various etiologies. The registry largely consisted of females, found a mean age of PAH presentation of 36 years, and was primarily Caucasian. PAH-specific therapies were not available at this time, and registry participants had a median survival of 2.8 years (1 year, 68%; 3 year, 48%; and 5 year, 34%).5,18 Significant progress in the field of PAH pathophysiology and treatment have occurred in the 2 decades since the NIH registry. Contemporary PAH registries vary in their study populations, study design, and cohorts. The 2002 French Network on Pulmonary Arterial Hypertension (French PAH) registry included 674 people with PAH.19 The French PAH registry found an estimated survival rate among those with idiopathic/familial/anorexigen-associated PAH of 82.9% at 1 year and 58.2% at 3 years.20 This is a markedly higher survival rate compared with the NIH registry but consistent with other more recent PAH registry findings.5,18,21 REVEAL (Registry to Evaluate Early and Long-Term PAH Disease Management) is a contemporary, multicenter, observational, US-based registry that began in 2006.22 In contrast to the NIH registry, REVEAL was specifically designed to ascertain demographics, longitudinal clinical course, and management of PAH from a current US perspective.21,23 Baseline characteristics for the 2967 individuals who met traditional hemodynamic criteria included a mean age of 53 (±14 years) and female sex in 79.5% with a female-to-male ratio of 4.8:1.24 In addition, 46% of individuals had idiopathic PAH, 25% were associated with connective tissue diseases, and 10% were associated with congenital heart diseases. There was also a mean duration between symptom onset and diagnosis of 2.8 years.25 Results of the REVEAL Registry showed a 1-year survival rate of 91% among 2716 individuals who were enrolled consecutively.26 An additional analysis assessing long-term survival (N = 2635, between March 2006 and December 2009) found survival rates of 85% at year 3, 68% at year 5, and 49% at year 7 from time of diagnosis.19 These increases in survival were attributed to various factors, including changes in treatment, increased patient support, and potentially, a difference in the PAH population cohort. Pathogenesis The triggering etiology that initiates the pathogenesis of PAH is likely multifactorial, including inappropriate angiogenesis, metabolic derangements, DNA damage, genetic mutations, and impaired vasoreactivity.3 Endothelial cell injury along with impaired vascular regeneration, abnormal vascular remodeling, and loss of the small pulmonary arteries are all known to occur as part of the PAH pathogenesis.27,28 Once endothelial dysfunction occurs, progressive vascular remodeling of the distal pulmonary arteries ensues, causing significant proliferation and resistance to apoptosis of pulmonary artery resident cells.3 As a result, pulmonary vascular lumen occlusion occurs, leading to increased pulmonary vascular resistance (PVR) and mPAP.3,29 The abnormal PVR and mPAP lead to right ventricle dilation and dysfunction, which can ultimately lead to a decreased cardiac output (CO). Additionally, an imbalance exists between vasodilation and vasoconstriction favoring vasoconstriction with an increase in circulating vasoconstrictors (ie, thromboxane, endothelin, and serotonin) and a decrease in circulating vasodilators (ie, prostacyclin, nitric oxide [NO], and vasoactive intestinal polypeptide).5 The improved understanding of the pathophysiology of PAH has led to development of therapies targeting the NO pathway, prostacyclin pathway, and endothelin pathway (Figure30).The pathology of PAH had previously been thought to be limited to the small pulmonary arteries; however, recent evidence suggests that systemic vascular manifestations also occur.31 For example, patients with PAH exhibit impaired brachial artery flow-mediated dilation, abnormal cerebral blood flow, skeletal myopathy, and intrinsic kidney disease. Although some of these manifestations can be explained as a consequence of right ventricular dysfunction, Nickel et al argue that there is also evidence to support a mechanistic link with PAH pathophysiology. More research is needed to fully understand the systemic effects of PAH.31 Tragically, as the disease progresses,thecompensatory mechanisms of the right heart can fail, and lead to premature death.3 Autoantibodies, proinflammatory cytokines, and inflammatory infiltrates have also been implicated in the pathogenesis of PAH. Individuals with PAH have increased von Willebrand factor levels, plasma fibrinopeptide A, plasminogen activator inhibitor-1, serotonin (5-HT), and thromboxane.32 In addition, tissue plasminogen activator, thrombomodulin, NO, and PGI2 are decreased, creating an imbalance that favors thrombosis.32 Genetics Over the past 20 years, there have been significant advancements in the understanding of the genetics of PAH.33,34 Approximately 6% to 10% of persons with PAH have a family history.35 Those who were previously identified as having idiopathic sporadic PAH are now known to have a genetic cause and would fall into the heritable PAH group instead. The most well-known genetic mutation associated with PAH is the bone morphongenetic protein receptor 2 (BMPR2) mutation. Seventy percent to 80% of patients with heritable PAH and 10% to 20% of individuals with idiopathic PAH have the BMPR2 mutation.35 Presence of this genetic mutation markedly increases the risk of developing heritable PAH and guidelines recommend genetic counseling for families and patients.36-38 Additional mutations that are implicated in PAH development involve ligands of BMPR2 and include GDF2 (encoding BMP9), type I receptor (ACVRL1), and SMAD9 (encoding Smad8). Potassium channel subfamily K member 3 (KCNK3) mutations and caveloin-1 (CAV1) mutations have also been identified along with many others.5 Interestingly, PAH mutations are autosomal dominant with low penetrance. As a result, some patients may never exhibit disease, further supporting the assertion that the trigger of disease is multifactorial.1 Mitochondrial metabolism impairments have also been associated with development of PAH.36 The female preponderance in PAH may be explained by the link between sex hormone metabolism differences and right ventricular function.36 More research is needed to elucidate this theory entirely. BMPR2 mutation penetrance also varies significantly, 14% of men and 42% of women, further suggesting that sex hormones and their metabolism may be associated with the pathogenesis of PAH.1 Clinical Presentation and Diagnosis A thorough history, physical examination, and complete workup is imperative to determine if a patient truly has PAH. The first presenting symptoms include exertional dyspnea, fatigue, and weakness.7,39,40 As the disease progresses, dyspnea may occur at rest, and other symptoms such as chest pain, presyncope, syncope, lower extremity edema, jugular venous distension, and abdominal bloating and distension may occur.7 Less common symptoms include cough, hemoptysis, and hoarseness. The assessment of symptoms includes placing the patient in a World Health Organization functional class (WHO-FC) based on level of impairment in physical activity. Patients in WHO-FC I have no limitation of physical activity; WHO-FC II is characterized by slight limitation in physical activity with ordinary activity causing undue dyspnea, fatigue, chest pain, or near syncope; WHO-FC III is characterized by marked limitation of physical activity with no discomfort at rest but less than ordinary physical activity causing undue dyspnea, fatigue, chest pain, or near syncope; finally, WHO-FC IV is characterized by an inability to perform any physical activity without symptoms with signs of right ventriculat failure and symptoms at rest with discomfort increasing by any physical activity. In addition to a thorough history and physical examination, initial tests such as chest radiography and electrocardiography should be done. If findings from the workup or clinical findings suggest the presence of PH and right ventricular dysfunction, a 2-dimensional transthoracic echocardiography (TTE) with doppler should be employed as an initial screening measure. TTE is the best test to screen for possible PH and PAH, but only a right heart catheterization (RHC) can assess the pulmonary hemodynamics needed to diagnose PH and PAH. The RHC is required to assess the mPAP, PVR, and CO. It is the gold standard and used to confirm a diagnosis of PH.7,40 Historically, PAH has been defined by an mPAP of greater than or equal to 25 mm Hg at rest plus a pulmonary wedge pressure (WP) less than or equal to 15 mm Hg, and PVR greater than or equal to 3 Wood units (WU) using RHC.41 However, at the 6th WSPH, the expert task force recommended to lower the hemodynamic definition for the first time since the inception of the WSPH in 1973 based on accumulated evidence suggesting a normal resting mPAP of 14 ± 3.3 mm Hg and that the upper limit of normal (or 2 standard deviations) for mPAP is greater than 20 mm Hg.1,42 The change in hemodynamic criteria was primarily driven by increasing evidence suggesting that those who fall within the 20 to 24 mm Hg mPAP range exhibited poorer outcomes and tended to progress to overt PH (especially those with systemic sclerosis, chronic thromboembolic pulmonary hypertension, and family history of PAH-causing genes) more often than those with an mPAP less than or equal to 20 mm Hg.43-47 The change in hemodynamic criteria has not occurred without opposition by some experts who argue that the 2 standard deviation argument is not consistent because pulmonary arterial WP and PVR cutoff values do not follow the same criteria. They suggest that the new hemodynamic criteria could create yet another cohort of individuals who could be "missed" by the previous and current cutoffs.48 Still, the evidence is clear that an mPAP greater than 20 mm Hg is considered above normal, and more research is required in patient cohorts to further elucidate the relationship between clinical presentation and long-term outcomes.1 Risk Stratification PAH treatment is based on the severity of disease at diagnosis and assessing how the individual will respond to treatment using multiple factors to stratify risk based on predicted mortality.1 Other treatment considerations include patient/clinician preference, drug interactions, tolerability, and potential adverse effects.49 Several risk stratification tools (RSTs) have been developed using retrospective analysis of large patient registries to aid in determining prognosis and guiding therapy for patients with PAH (Table 349). The RSTs use multiple data points such as demographics, functional status, laboratory values, and hemodynamic information to stratify patients into low, intermediate, or high risk. The categories are then used as a baseline for initiating treatment, determining prognosis, and monitoring response and disease progression long term.1,48 The REVEAL RST consists of 12 to 14 variables used to determine the risk of 1-year mortality. The RST has been validated for predicting survival at baseline, 1-year follow-up, and at 5 years.19,48,50,51 The most recent REVEAL 2.0 calculator can also predict clinical worsening and mortality among those who have survived PAH for a minimum of 1 year from their initial enrollment by including hospitalizations in the past 6 months and estimated glomerular filtration rate.1,48 Another RST, COMPERA, was developed by a European group and uses fewer data points than REVEAL but classifies individuals similarly. The 2015 European Society of Cardiology (ESC)/European Respiratory Society (ERS) PH guidelines RST uses a multidimensional approach that focuses on the most frequent determinants of prognosis.39 All of the variables do not have to be assessed at each visit, but should include FC determination and at least one exercise capacity measurement (eg, 6-minute walking distance). It is also recommended to assess RV function by measuring brain natriuretic peptide (BNP)/N-terminal pro-brain BNP (NT-proBNP) or by echocardiography. Patients are then stratified into low, intermediate, and high risk based on determinants assessed. The RST can assist in guiding therapeutic decisions; however, application to individual patients should be done with care. A comprehensive assessment that also includes other risk factors such as signs of right heart failure, syncope, and comorbidities should be included to optimize clinical decision making.39 The Swedish PAH Register and the French Pulmonary Hypertension Network have also developed RSTs from large registries. Regardless of which RST is used, all have similar efficacy in identifying individuals at high risk. It is important to note that the tools utilize retrospective data and have some limitations, including measurement of nonmodifiable risk factors and inclusion of data points that are not routinely collected in PAH. Thus, despite which tool is selected, RSTs can help clinicians determine which individuals with PAH are at high risk for 1-year mortality, prioritization of therapies, and referral for transplant.48 Quality of Life The impact of PAH disease symptoms on a person's functional mobility and psychosocial state adversely affects health-related quality of life (HRQOL).52 Although there have been significant advancements in the understanding of PAH and targeted therapies that have decreased mortality, these improvements have not necessarily been paralleled from the perspective of individuals with PAH.1 PAH affects all parts of a person's daily life that influence their HRQOL, including physical activity, well-being, and emotional and social functioning.52 The debilitation level experienced by patients is considered at least as severe as chronic obstructive pulmonary disease and renal failure.52 A report from the European Pulmonary Hypertension Association (PHA Europe) found that 83% (n = 326) of people with PAH surveyed reported difficulty climbing stairs, and 97% stated PAH affected their ability to participate in sports and exercise to some degree.52 Similarly, the FDA Patient Voice survey (≈85 participants) reported that breathlessness and fatigue were also restrictive on daily physical activities. Both surveys also found that reduced physical activity had negative implications for long-term outcomes among persons with PAH.52 PAH also has considerable psychological effects, such as feelings of social isolation, lack of understanding or knowledge about the disease in the community (not a "visible" disease), and friends and family.53 A study also found that 48% of individuals with PAH experienced mild to extremely severe symptoms of anxiety, 32.6% had symptoms of depression, and 27.6% had symptoms of stress.54 As the psychological impact of PAH is often underrecognized, it is vital to assess patients during all encounters, especially if and when functional class worsens. In addition, patients with PAH are also frequently affected by a loss of household income due to loss of work or inability to remain working.53 A European PHA survey found that 73% of patients who had to give up work or needed assistance to maintain employment had a loss in average household income. Moreover, 1 in 6 reported their income decreasing by half. Also, 35% of caregivers reported a reduction in income to care for the individual with PAH.53 Age has contributed to the burden of patients with PAH.55 The prevalence of PAH is increasing among people aged 50 to 65 years who are more likely to be diagnosed with advanced stages of disease, and have lower exercise capacity and a higher number of comorbidities. Multiple comorbidities are associated with a delay in diagnosis among older patients and could explain the challenge of disease recognition in earlier years among this patient population.55 Women of childbearing age are also at increased risk of complications during pregnancy due to poor tolerance of hemodynamic and physiologic changes that can cause right ventricular failure and arrhythmias.56 Due to a significantly higher associated mortality rate, it is recommended that women with PAH avoid pregnancy.39 Multiple factors contribute to an individual's perception of their overall well-being. Therefore, the WSPH expert task force recommends that management of individuals with PH should occur at specialized care centers with a patient-centered multidisciplinary team that focuses on quality of life, shared clinical decision making, and access to palliative care. Although numerous studies have demonstrated that PH treatments improve HRQOL, it is important to recognize that HRQOL is one component and may not capture the depth and complexity of psychosocial issues experienced by patients and caregivers. In other words, both individual and patient population-level perspectives should be considered.1 Several patient-reported outcome (PRO) instruments have been developed to evaluate the effect of PAH therapies on patient symptoms and the impact of the symptoms on patients' lives and use of these are recommended to be incorporated as secondary end points in clinical trials. Two of these instruments are the psychometric validation of the pulmonary arterial hypertension-symptoms and impact (PAH-SYMPACT) questionnaire and the emphasis-10 questionnaire.57,58 The WSPH task force encourages clinicians to participate in narrative medicine where individuals with PAH can express their concerns and challenges related to their individual health.1 Patients with PAH also need improved access to palliative care, as they may experience low HRQOL and high disease burden.59 Concerning the population level, continued support for patient groups and associations, patient education, and public awareness are needed.1 The interdisciplinary healthcare team can help patients navigate intolerable adverse effects and make recommendations for treatment adjustments and avoidance of drug interactions as required to optimize therapy and treatment acceptance and/or adherence. Conclusions PAH is a devastating life-limiting progressive disorder. Over the past 20 years, significant advancements have occurred due to an improved understanding of PAH pathogenesis and specific therapies that help decrease mortality. As more evidence has accumulated, changes to the evaluation and management of PAH have occurred. Contemporary registries continue to provide crucial information to help risk-stratify patients, determine prognosis, and monitor and manage therapeutic goals. Patients with PAH experience significant effects on their HRQOL, which is correlated to their functional, emotional, work, and social abilities. Healthcare providers should assess a patient's HRQOL during each encounter to improve patient satisfaction. Furthermore, as the factors that affect individual HRQOL are multifactorial, it is important that patients are involved in the clinical decision-making process. A multidisciplinary approach with multiple layers of support should be available to all patients, and importantly, they must be aware of the existence of such services. Author affiliation: Deborah Jo Levine, MD, FCCP, is professor of medicine, pulmonary and critical care, and director of pulmonary hypertension, University of Texas Health, San Antonio, TX. Funding source: This activity is supported by an educational grant from United Therapeutics Corporation. Author disclosure: Dr Levine has no relevant financial relationships with commercial interests to disclose. Authorship information: Substantial contributions to concept and design; drafting of the manuscript; and critical revision of the manuscript for important intellectual content. Address correspondence to: levinedj@uthscsa.edu Medical writing and editorial support: Brittany Hoffmann-Eubanks, PharmD, MBA REFERENCES 1. Beshay S, Sahay S, Humbert M. Evaluation and management of pulmonary arterial hypertension. Respir Med. 2020;171:106099. doi: 10.1016/j.rmed.2020.106099 2. Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 suppl):D34-D41. doi: 10.1016/j.jacc.2013.10.029 3. Bourgeois A, Omura J, Habbout K, et al. Pulmonary arterial hypertension: new pathophysiological insights and emerging therapeutic targets. Int J Biochem Cell Biol. 2018;104:9-13. doi: 10.1016/j.biocel.2018.08.015 4. Klinger JR, Elliott CG, Levine DJ, et al. Therapy for pulmonary arterial hypertension in adults: update of the CHEST Guideline and Expert Panel Report [Erratum appears in Chest. 2021;159(1):457]. Chest. 2019;155(3):565-586. doi: 10.1016/j.chest.2018.11.030 5. Prins KW, Thenappan T. World Health Organization group I pulmonary hypertension: epidemiology and pathophysiology. Cardiol Clin. 2016;34(3):363-374. doi: 10.1016/j.ccl.2016.04.001 6. Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913. doi: 10.1183/13993003.01913-2018 7. McLaughlin VV, Shah SJ, Souza R, Humbert M. Management of pulmonary arterial hypertension. J Am Coll Cardiol. 2015;65(18):1976-1997. doi: 10.1016/j.jacc.2015.03.540 8. Zamanian RT, Hedlin H, Greuenwald P, et al. Features and outcomes of methamphetamine- 9. Hickey PM, Thompson AA, Charalampopoulos A, et al. Bosutinib therapy resulting in severe deterioration of pre-existing pulmonary arterial hypertension. Eur Respir J. 2016;48(5):1514-1516. doi: 10.1183/13993003.01004-2016 10. Renard S, Borentain P, Salaun E, et al. Severe pulmonary arterial hypertension in patients treated for hepatitis C with sofosbuvir. Chest. 2016;149(3):e69-e73. doi: 10.1016/j.chest.2015.09.018 11. Riou M, Seferian A, Savale L, et al. Deterioration of pulmonary hypertension and pleural effusion with bosutinib following dasatinib lung toxicity. Eur Respir J. 2016;48(5):1517-1519. doi: 10.1183/13993003.01410-2016 12. Seegobin K, Babbar A, Ferreira J, Lyons B, Cury J, Seeram V.. A case of worsening pulmonary arterial hypertension and pleural effusions by bosutinib after prior treatment with dasatinib. Pulm Circ. 2017;7(4):808-812. doi: 10.1177/2045893217733444 13. Alvarez PA, Saad AK, Flagel S, Mazzocchi O, Blanco MV. Leflunomide-induced pulmonary hypertension in a young woman with rheumatoid arthritis: a case report. Cardiovasc Toxicol. 2012;12(2):180-183. doi: 10.1007/s12012-012-9153-3 14. Coirier V, Lescoat A, Chabanne C, et al. Pulmonary arterial hypertension in four patients treated by leflunomide. Joint Bone Spine. 2018;85(6):761-763. doi: 10.1016/j.jbspin.2017.12.014 15. Savale L, Chaumais MC, Montani D, et al. Direct-acting antiviral medications for hepatitis C virus infection and pulmonary arterial hypertension. Chest. 2016;150(1):256-258. doi: 10.1016/j.chest.2016.04.031 16. Mandel J, Mark EJ, Hales CA. Pulmonary veno-occlusive disease. Am J Respir Crit Care Med. 2000;162(5):1964-1973. doi: 10.1164/ajrccm.162.5.9912045 17. Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327(2):76-81. doi: 10.1056/nejm199207093270203 18. Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107(2):216-223. doi: 10.7326/0003-4819-107-2-216 19. Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest. 2012;142(2):448-456. doi: 10.1378/chest.11-1460 20. Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122(2):156-163. doi: 10.1161/circulationaha.109.911818 21. Benza RL, Gomberg-Maitland M, Miller DP, et al. The REVEAL Registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest. 2012;141(2):354-362. doi: 10.1378/chest.11-0676 22. McGoon MD, Miller DP. REVEAL: A contemporary US pulmonary arterial hypertension registry. Eur Respir Rev. 2012;21(123):8-18. doi: 10.1183/09059180.00008211 23. McGoon MD, Krichman A, Farber HW, et al. Design of the REVEAL registry for US patients with pulmonary arterial hypertension. Mayo Clin Proc. 2008;83(8):923-931. doi: 10.4065/83.8.923 24. Frost AE, Badesch DB, Barst RJ, et al. The changing picture of patients with pulmonary arterial hypertension in the United States: how REVEAL differs from historic and non-US contemporary registries. Chest. 2011;139(1):128-137. doi: 10.1378/chest.10-0075 25. Badesch DB, Raskob GE, Elliott CG, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest. 2010;137(2):376-387. doi: 10.1378/chest.09-1140 26. Benza RL, Miller DP, Gomberg-Maitland M, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. 2010;122(2):164-172. doi: 10.1161/circulationaha.109.898122 27. Ranchoux B, Harvey LD, Ayon RJ, et al. Endothelial dysfunction in pulmonary arterial hypertension: an evolving landscape (2017 Grover Conference Series). Pulm Circ. 2018;8(1):2045893217752912. doi: 10.1177/2045893217752912 28. Yuan JX, Rubin LJ. Pathogenesis of pulmonary arterial hypertension: the need for multiple hits. Circulation. 2005;111(5):534-538. doi: 10.1161/01.CIR.0000156326.48823.55 29. Humbert M, Guignabert C, Bonnet S, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. 2019;53(1):1801887. doi: 10.1183/13993003.01887-2018 30. Humbert M, Ghofrani HA. The molecular targets of approved treatments for pulmonary arterial hypertension. Thorax. 2016;71(1):73-83. doi: 10.1136/thoraxjnl-2015-207170 31. Nickel NP, Yuan K, Dorfmuller P, et al. Beyond the lungs: systemic manifestations of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2020;201(2):148-157. doi: 10.1164/rccm.201903-0656CI 32. Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol. 2011;8(8):443-455. doi: 10.1038/nrcardio.2011.87 33. Deng Z, Morse JH, Slager SL, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67(3):737-744. doi: 10.1086/303059 34. Lane KB, Machado RD, Pauciulo MW, et al; International PPH Consortium. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26(1):81-84. doi: 10.1038/79226 35. Morrell NW, Aldred MA, Chung WK, et al. Genetics and genomics of pulmonary arterial hypertension. Eur Respir J. 2019;53:1801899. doi: 1183/13993003.0189-2018 36. Evans JD, Girerd B, Montani D, et al. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. Lancet Respir Med. 2016;4(2):129-137. doi: 10.1016/S2213-2600(15)00544-5 37. Loyd JE, Primm RK, Newman JH. Familial primary pulmonary hypertension: clinical patterns. Am Rev Respir Dis. 1984;129(1):194-197. doi: 10.1164/arrd.1984.129.1.194 38. Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Rev Esp Cardiol (Engl Ed). 2016;69(2):177. doi: 10.1016/j.rec.2016.01.002 39. Galiè N, Humbert M, Vachiery JL, et al; ESC Scientific Document Group. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37(1):67-119. doi: 10.1093/eurheartj/ehv317 40. Frost A, Badesch D, Gibbs JS, et al. Diagnosis of pulmonary hypertension. Eur Respir J. 2019;53(1):1801904. doi: 10.1183/13993003.01904-2018 41. Condon DF, Nickel NP, Anderson R, Mirza S, de Jesus Perez VA. The 6th World Symposium on Pulmonary Hypertension: what's old is new. F1000Res. 2019;8:F1000. doi: 10.12688/f1000research.18811.1 42. Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J. 2009;34(4):888-894. doi: 10.1183/09031936.00145608 43. Maron BA, Hess E, Maddox TM, et al.Association of borderline pulmonary hypertension with mortality and hospitalization in a large patient cohort: insights from the Veterans Affairs clinical assessment, reporting, and tracking program. Circulation. 2016;133(13):1240-1248. doi: 10.1161/CIRCULATIONAHA.115.020207 44. Assad TR, Maron BA, Robbins IM, et al. Prognostic effect and longitudinal hemodynamic 45. Douschan P, Kovacs G, Avian A, et al. Mild elevation of pulmonary arterial pressure as a predictor of mortality. Am J Respir Crit Care Med. 2018;197(4):509-516. doi: 10.1164/rccm.201706-1215OC 46. Valerio CJ, Schreiber BE, Handler CE, Denton CP, Coghlan JG. Borderline mean pulmonary artery pressure in patients with systemic sclerosis: transpulmonary gradient predicts risk of developing pulmonary hypertension. Arthritis Rheum. 2013;65(4):1074-1084. doi: 10.1002/art.37838 47. Coghlan JG, Wolf M, Distler O, et al. Incidence of pulmonary hypertension and determining factors in patients with systemic sclerosis. Eur Respir J. 2018;51(4):1701197. doi: 10.1183/13993003.01197-2017 48. Thomas CA, Anderson RJ, Condon DF, de Jesus Perez VA. Diagnosis and management of pulmonary hypertension in the modern era: insights from the 6th World Symposium. Pulm Ther. 2020;6(1):9-22. doi: 10.1007/s41030-019-00105-5 49. Galiè N, Channick RN, Frantz RP, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. 2019;53(1):1801889. doi: 10.1183/13993003.01889-2018 50. Benza RL, Miller DP, Foreman AJ, et al. Prognostic implications of serial risk score assessments in patients with pulmonary arterial hypertension: a Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) analysis. J Heart Lung Transplant. 2015;34(3):356-361. doi: 10.1016/j.healun.2014.09.016 51. Farber HW, Miller DP, Poms AD, et al. Five-year outcomes of patients enrolled in the REVEAL Registry. Chest. 2015;148(4):1043-1054. doi: 10.1378/chest.15-0300 52. Delcroix M, Howard L. Pulmonary arterial hypertension: the burden of disease and impact on quality of life. Eur Respir Rev. 2015;24(138):621-629. doi: 10.1183/16000617.0063-2015 53. European Pulmonary Hypertension Association. The impact of pulmonary arterial hypertension (PAH) on the lives of patients and carers: results from an international survey. Published September 2012. Accessed January 8, 2021. phaeurope.org/wp-content/uploads/PAH_Survey_FINAL.pdf 54. M M Vanhoof J, Delcroix M, Vandevelde E, et al. Emotional symptoms and quality of life in patients with pulmonary arterial hypertension. J Heart Lung Transplant. 2014;33(8):800-808. doi: 10.1016/j.healun.2014.04.003 55. Hoeper MM, Gibbs JS. The changing landscape of pulmonary arterial hypertension and implications for patient care. Eur Respir Rev. 2014;23(134):450-457. doi: 10.1183/09059180.00007814 56. Tabarsi N, Levy R, Rychel V, et al. Pregnancy among women with pulmonary arterial hypertension: a changing landscape? Int J Cardiol. 2014;177(2):490-491. doi: 10.1016/j.ijcard.2014.08.059 57. Chin KM, Gomberg-Maitland M, Channick RN, et al. Psychometric validation of the Pulmonary Arterial Hypertension-Symptoms and Impact (PAH-SYMPACT) Questionnaire: results of the SYMPHONY trial. Chest. 2018;154(4):848-861. doi: 10.1016/j.chest.2018.04.027 58. Yorke J, Corris P, Gaine S, et al. emPHasis-10: development of a health-related quality of life measure in pulmonary hypertension. Eur Respir J. 2014;43(4):1106-1113. doi: 10.1183/09031936.00127113 59. Swetz KM, Shanafelt TD, Drozdowicz LB, et al. Symptom burden, quality of life, and attitudes toward palliative care in patients with pulmonary arterial hypertension: results from a cross-sectional patient survey. J Heart Lung Transplant. 2012;31(10):1102-1108. doi: 10.1016/j.healun.2012.08.010 |
| Right-Sided Heart Failure: Symptoms, Causes, and Treatments - Healthline Posted: 04 May 2021 12:00 AM PDT  Heart failure is a general term for a weakening of the heart muscle that leaves it unable to pump enough blood to meet the demands of the body. There are actually several types of heart failure, each with its own cause and complications. Right-sided heart failure involves the part of the heart responsible for pumping blood to the lungs, where it receives oxygen. The blood then travels throughout the body to deliver oxygen to your organs, muscles, and other tissues. While treatment options vary, they usually involve a comprehensive approach to maintaining the health of your entire heart and circulatory system. The outlook for someone with right-sided heart failure depends on the severity of the disease, as well as how early treatment begins. While it's sometimes a life threatening condition, it can be managed with a combination of medications, lifestyle changes, and in some cases, surgery. The term heart failure sounds like the heart has stopped pumping, similar to how the phrase "engine failure" means an engine no longer produces any power. Heart failure actually means that the heart muscle has grown weaker and can no longer provide sufficient blood flow to all parts of the body. The heart still pumps, just not as efficiently and effectively as it once did. As a result, the body doesn't get all the oxygenated blood it needs to function and complications affecting various other organs can develop. What is left-sided heart failure?Left-sided heart failure is a more common condition than right-sided heart failure. It happens when the left ventricle has to pump harder than usual to try to deliver enough blood to keep the body healthy. There are two types of left-sided heart failure:
Fluid retention causing swelling in the lower limbs and sometimes the abdomen is a common and obvious symptom of right-sided heart failure, but there are several other symptoms that may develop: Several factors can weaken the heart and trigger heart failure. Conditions that damage your heart, such as a heart attack, or that make your heart work harder, such as valve disease, can have the same result. Right-sided heart failure can also be brought on by lung disease or pulmonary hypertension. Right-sided heart failure is most commonly brought on by left-sided heart failure. When the left side of your heart weakens, blood can build up within the chambers. This increases the pressure within the blood vessels carrying blood to the lungs — a condition known as pulmonary hypertension. To compensate, the right side of the heart must work harder. Eventually, the right side weakens from the extra effort, and you develop right-sided heart failure. Right-sided heart failure can also be a result of leaky or damaged right-sided valves, such as a leaky tricuspid valve (tricuspid regurgitation). Specific risk factors for right-sided heart failure include:
The proper treatment for right-sided heart failure depends on the underlying condition causing it. Treating right-sided heart failure usually involves the use of one or more medications, lifestyle measures, and possibly implanted devices that support the heart's ability to pump. MedicationsThe following types of medications are among those commonly prescribed to treat right-sided heart failure:
LifestyleTo help your heart work efficiently, the following lifestyle measures are important:
DevicesFor more serious cases of right-sided heart failure, you may need an implanted device to support healthy heart function. A mechanical heart pump can take the form of a ventricular assist device or a total artificial heart, to compensate for the heart's loss of pumping strength. In some cases, surgery may be needed to correct a congenital heart defect that caused the heart failure. Or, in the most serious cases, a heart transplant may be necessary if other treatment options have been unsuccessful. Right-sided heart failure is a lifelong condition, and there is currently no cure. However, many people manage symptoms and maintain a decent quality of life. The key is to work closely with your doctor and follow your medication regimen exactly as prescribed. It's also critical to report any new symptoms and to manage any other medical conditions that could contribute to or worsen because of heart failure. These may include: There are advances in mechanical support devices, suggesting that treatment options will continue to save and extend lives. |
| Right sided heart failure: Symptoms, outlook, treatment - Medical News Today Posted: 20 May 2021 12:00 AM PDT 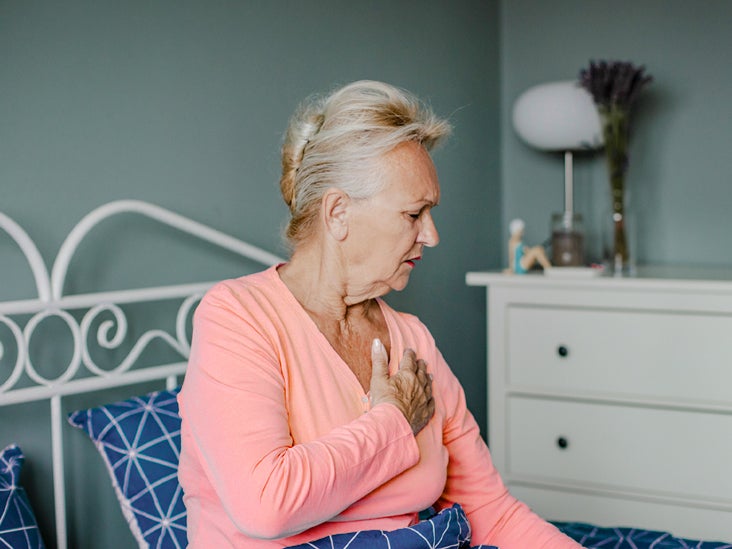 If a person has right sided heart failure, it means the right side of their heart is not pumping blood to the lungs as effectively. The condition can develop if a person has already experienced weakness in the left side of the heart. Experts also refer to right sided heart failure as pulmonary heart disease. In most cases, right sided heart failure results from problems that have already occurred in the left ventricle. However, it can also develop if a person is experiencing generalized heart failure or lung disease. Blood can become blocked in the left ventricle and lungs. This puts extra stress on the right ventricle to pump the blood into the lungs. Over time, the right side of the heart can weaken and start to fail. This article will outline the characteristics of right sided heart failure. It will also discuss its symptoms and causes and treatment options. When a person experiences heart failure, it means the heart muscle is not strong or flexible enough to pump blood throughout the body. The right side of the heart takes deoxygenated blood that has already been through the heart and pumps it out into the lungs. The lungs then replenish it with oxygen. In right sided heart failure, the right ventricle of the heart is unable to pump enough blood to the lungs. This often happens due to failure in the left side of the heart. Increased fluid and pressure then pass through the right side of the heart muscle and into the lungs. Consequently, the right side of the heart becomes weaker and damaged. There is also a buildup of blood in the veins, which can cause dispersion of fluid to surrounding tissues. This in turn can lead to swelling throughout the body, including the:
When right sided heart failure occurs, the increased pressure in the veins causes dispersion of fluid to surrounding tissues. This happens because this side of the heart is too weak to pump blood forward to the lungs. The result may be a buildup of fluid in various parts of the body, including the legs, abdomen, and liver. By contrast, when left sided heart failure occurs, it means the left ventricle is not pumping enough blood throughout the body. Blood then accumulates in the pulmonary veins, which are blood vessels that carry blood away from the lungs. When left sided heart failure occurs, a person can experience the following: While left sided heart failure is the most common type of heart failure, it can lead to right sided heart failure. A person with isolated right sided heart failure may experience the following: Right sided heart failure can be due to the following conditions: Left sided heart failureLeft sided heart failure is the primary cause of right sided heart failure. When the left ventricle is not working as effectively, fluid pressure increases and ends up moving back through the lungs. This can cause an overload to the heart's right side. Consequently, when the right side is unable to pump blood, fluid accumulates in the veins, resulting in swelling. Pulmonary hypertensionRight sided heart failure is the main consequence of pulmonary hypertension, which is when there is high blood pressure in the blood vessels that deliver oxygen-rich blood to the lungs. If there are changes in the small blood vessels inside the lungs, high blood pressure can occur in the right side of the heart. As a result of the added strain, the heart has difficulty pumping blood to the lungs. Chronic conditions such as pulmonary embolism and chronic obstructive pulmonary disease (COPD) and diseases that cause difficulty breathing, including chronic bronchitis and emphysema, can cause strain on the right side of the heart. Congenital heart conditionsAbnormal heart function can result from structural heart conditions that are present at birth. Conditions may include:
These conditions may affect how blood flows through the heart and to the rest of the body. Pulmonic stenosisA person with pulmonic stenosis experiences a narrowing of the pulmonic valve. This decreases blood flow out from the right ventricle, which then needs to work harder. Similar to chronic lung disease, this extra strain can cause the right ventricle to fail over a period of time. Other possible causesOther causes of right sided heart failure include:
To diagnose right sided heart failure, a cardiologist, who specializes in the treatment of disorders of the heart and blood vessels, will perform a thorough exam. This can include a medical history evaluation and other tests, such as:
Treatment for right sided heart failure aims to manage symptoms. Treating risk factors for pulmonary hypertension is an important step toward keeping this condition under control. Doctors will recommend treatment options depending on the cause of the condition. MedicationIf a person has right sided heart failure, a doctor may need to remove extra sodium and fluid and relax blood vessels. Medications will aim at: Behavioral changesA person with a right sided heart failure diagnosis may need to make certain behavioral changes, including:
Learn about how to follow a cardiac diet here. SurgeryIf medication and lifestyle changes prove ineffective, a doctor may recommend a transplant of a lung, the heart, or both. A person with right sided heart failure will likely need treatment for the rest of their life. While there is currently no cure, there are steps a person can take to manage symptoms and treat the cause of their heart failure. Chronic right sided heart failure can be a result of a number of conditions, including left sided heart failure. A person with a right sided heart failure diagnosis should consult a doctor to find the most suitable management and treatment options. Researchers are working to develop new therapies. However, treatment may last for the rest of a person's life. Making lifestyle changes, such as reducing stress and avoiding alcohol, can help reduce symptoms. It is also important to manage contributing health factors, such as blood pressure and anemia. |
| You are subscribed to email updates from "pulmonary hypertension causes" - Google News. To stop receiving these emails, you may unsubscribe now. | Email delivery powered by Google |
| Google, 1600 Amphitheatre Parkway, Mountain View, CA 94043, United States | |


Comments
Post a Comment