Evolving management and treatment of pulmonary hypertension ... - Dove Medical Press
Introduction
Pulmonary hypertension (PH) is defined as a mean pulmonary artery pressure (PAP) greater than or equal to 20 mmHg as measured by right heart catheterization.1,3 Several mechanisms behind the development of elevated pulmonary artery pressures have led to the World Health Organization (WHO) classification of pulmonary hypertensive diseases into five distinct groups.1 One of the foremost challenges in approaching a patient with pulmonary hypertension is the task of correctly diagnosing and classifying the disease in order to determine appropriate treatment. In some cases, optimal management of an associated disease is the primary treatment of choice, but in other situations targeted medications or surgical measures may be indicated. Over the last two decades, a substantial scientific effort has been invested in understanding the pathobiology behind WHO Group 1 pulmonary arterial hypertension (PAH). PAH is a disease affecting the pulmonary arteries and characterized by specific changes in arterial morphology and rising pulmonary vascular resistance that can lead to right heart failure and death. Pathophysiologic pathways involved in PAH have been defined.2,3 A number of novel mechanisms and mediators are under investigation. The discovery of specific disease-causing mechanisms has fostered development of targeted therapies that are effective in controlling the progressive arteriopathy and symptoms of PAH. Several investigations are underway to elucidate if these same targeted modifiers might be beneficial in the treatment of pulmonary hypertension in other WHO Groups. This review will describe the basic mechanisms involved in the classification of pulmonary hypertensive diseases, elaborate in more detail on the state-of-the-art understanding of PAH pathobiology, outline current management strategies to achieve optimal outcomes for patients with PAH, and summarize more recent efforts to identify solutions for the treatment of PH in other WHO Groups.
Diagnosis and Classification of Pulmonary Hypertensive Diseases
The pulmonary hypertensive diseases were first classified by the World Health Organization according to pathological and clinical features in 1973 during the 1st World Symposium on Pulmonary Hypertension in Geneva, Switzerland.4 The WHO classification system has been further refined over time with the most recent updates coming from the 6th World Symposium in 2018 and the 2022 ESC/ERS (European Society of Cardiology/European Respiratory Society) Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension.1,3 Classification is based on the primary mechanisms causing elevated pulmonary arterial pressure (Figure 1). Patients with WHO Group 1 PAH have a distinct arteriopathy characterized by excessive proliferation of the cellular components of the vascular wall, smooth muscle hypertrophy, in situ thrombosis and formation of plexiform lesions that occlude the vessel lumen.5,6 Hyperproliferative changes that occur in this group may also develop in pulmonary venous structures. WHO Group 2 PH results primarily from rising postcapillary pressures as seen in diseases affecting the left side of the heart, such as cardiomyopathies or valvular heart disease. A third mechanism causing pulmonary hypertension is mediated by hypoxia and associated vasoconstriction. Hypoxic vasoconstriction in WHO Group 3 PH may be associated with high altitude, sleep apnea and other lung diseases, such as pulmonary fibrosis or emphysema. Circulatory flow obstruction, resulting from thromboembolic or other embolic events, can result in WHO Group 4 pulmonary hypertension. The final classification, WHO Group 5, consists of disorders that are associated with pulmonary hypertension, without any unifying mechanistic features.
 | Figure 1 Classification of pulmonary hypertensive diseases. Abbreviations: HF, heart failure; PAH, pulmonary arterial hypertension; PCH, pulmonary capillary hemangiomatosis; PH, pulmonary hypertension; PVOD, pulmonary veno-occlusive disease. Notes: aPatients with heritable PAH or PAH associated with drugs and toxins might be acute responders. bLeft ventricular ejection fraction for HF with reduced ejection fraction: <40%; for HF with mildly reduced ejection fraction: 41–49%. cOther causes of pulmonary artery obstructions might include: sarcoma (high or intermediate grade or angiosarcoma), other malignant tumors (eg, renal carcinoma, uterine carcinoma, germ-cell tumors of the testes), non-malignant tumors (eg, uterine leiomyoma), arteritis without connective tissue disease, congenital pulmonary arterial stenoses, and hydatidosis. dIncluding inherited and acquired chronic hemolytic anemia and chronic myeloproliferative disorders. eIncluding sarcoidosis, pulmonary Langerhan's cell histiocytosis, and neurofibromatosis type 1. fIncluding glycogen storage diseases and Gaucher disease. Reproduced with permission from Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2023 Jan 6;61(1):2200879. Copyright 2023 European Society of Cardiology & European Respiratory Society.3 |
The presence of pulmonary hypertension may be suspected from medical history, symptoms, and findings on electrocardiogram or chest radiographs.7 A careful history of other disease states is essential in determining causal relationships and classification according to WHO criteria. Connective tissue disease, portal hypertension, human immunodeficiency virus infection, congenital heart disease, and certain drug or toxin exposures have been associated with development of the same arteriopathy seen in PAH.8 Symptoms are nonspecific and overlap with those seen in many other cardiopulmonary diseases. Data from the REVEAL (Registry to Evaluate Early and Long-term Pulmonary Arterial Hypertension Management) registry indicate that shortness of breath and fatigue are the most common presenting symptoms. Chest pain is often noted but less prevalent. Edema and syncope are ominous symptoms that usually indicate a more advanced stage of pulmonary hypertension.9,10 Physical manifestations of pulmonary hypertension on exam may include an accentuated second pulmonic heart sound, a systolic murmur due to tricuspid regurgitation, and a right ventricular lift. Jugular venous distension, ascites and edema represent manifestations of advanced pulmonary vascular disease.11 Signs of right ventricular hypertrophy or right axis deviation on ECG are indicators of underlying pulmonary hypertension, as are enlarged central pulmonary arteries, peripheral vascular pruning and obliteration of the retrosternal clear space on chest radiographs.7,12
When history, exam findings and basic diagnostic studies suggest pulmonary hypertension, the suspicion can be confirmed by further screening with an echocardiogram.13–15 The standard transthoracic echocardiogram can provide an estimate of pulmonary artery pressure from velocity of tricuspid regurgitation. The echocardiogram is also helpful in distinguishing left heart causes for pulmonary hypertension and provides useful prognostic information from assessment of right ventricular characteristics and presence or absence of pericardial effusion. After echocardiography, the diagnostic algorithm includes a process of eliminating causes of secondary pulmonary hypertension (Figure 2). Current guidelines recommend an algorithmic approach which includes bloodwork to screen for autoimmune connective tissue disease, HIV infection, liver and thyroid dysfunction, screening for sleep apnea, CT scanning to exclude fibrotic or emphysematous parenchymal lung disease, a VQ scan to eliminate thromboembolic disease and pulmonary function testing to identify the presence of obstructive or restrictive lung disease.3,16
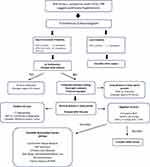 | Figure 2 Algorithm for the diagnosis of pulmonary arterial hypertension. Abbreviations: ECG, electrocardiogram; CXR, chest x-ray; VTR, velocity tricuspid regurgitation; LV, left ventricle; PH, pulmonary hypertension; PAH, pulmonary arterial hypertension; VQ, ventilation perfusion; CT, CT scan; RHC, right heart catheterization; mPAP, mean pulmonary artery pressure; CWP, capillary wedge pressure; PVR, pulmonary vascular resistance; WU, Wood Units; HIV, human immunodeficiency virus. Notes: Swisher JW andKailash S. Advances in management of pulmonary hypertension associated with systemic sclerosis. In: New Insights Into Systemic Sclerosis. (Michal Tomcik, ed.) InTech Open, London, UK.© 2019 The Author(s). Licensee IntechOpen. This chapter is distributed under the terms of the Creative Commons Attribution 3.0 License, which permits unrestricted use, distribution, and reproduction in any medium.17 |
Right heart catheterization provides the most accurate measurement of pulmonary arterial pressure and is the final confirmatory diagnostic study. A complete right heart study also determines vasoreactivity, evaluates post-capillary pulmonary circulatory pressure, identifies intracardiac left to right shunts and provides valuable prognostic data.18 The traditional hemodynamic definition of precapillary PAH was updated at the 6th World Symposium on Pulmonary Hypertension in 2018 to include: a) mean PA pressure >20 mmHg, b) pulmonary capillary wedge pressure ≤15 mmHg, and c) pulmonary vascular resistance (PVR) ≥3 WU (Wood Units).1 The recently released 2022 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension further modify this hemodynamic definition by lowering the PVR criterion to any value >2 WU.3 Figure 3 includes a summary of the most recent hemodynamic definitions for PH from the ESC/ERS Guidelines. In patients with idiopathic, heritable or drug-related PAH vasoreactivity testing identifies a small subset of PAH patients who may respond well to treatment with calcium blockers. Vasoreactivity testing can be performed using inhaled nitric oxide, adenosine or intravenous epoprostenol. A positive vasoreactivity study is determined by a decrease in mean PAP by at least 10 mmHg and an absolute value of mean PAP less than 40 mmHg.19 Adjunctive testing with exercise, straight leg raise or fluid challenge during right heart catheterization can unmask postcapillary pulmonary hypertension seen with heart failure and preserved ejection fraction.20–22 Certain data collected during right heart catheterization, in particular the right atrial pressure and cardiac index, have been shown to be important predictors of long-term survival.23,24
 | Figure 3 Hemodynamic definitions of pulmonary hypertension. Abbreviations: mPAP, mean pulmonary arterial pressure; PAWP, pulmonary arterial wedge pressure; PVR, pulmonary vascular resistance; WU, Wood Units. Notes: Reproduced with permission from Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2023 Jan 6;61(1):2200879. Copyright 2023 European Society of Cardiology & European Respiratory Society.3. |
Epidemiology
Most demographic data reported for pulmonary hypertension pertain to those patients with WHO Group 1 PAH. There has been little detailed information available about epidemiology in other WHO Groups. Perhaps, the most comprehensive report of epidemiology in WHO Groups 1 through 4 comes from a large population-based cohort study in Ontario, Canada.25 Data pertaining to the incidence, prevalence, comorbidities and mortality in this report were extracted from hospitalization, emergency room visit and universal coverage health service records for 50,529 residents of Ontario from 1993 to 2012. It is important to note that reliability of data in this report was dependent on the accuracy of diagnostic coding. Further, only 40.9% of incident patients in this cohort underwent a detailed diagnostic workup that included right heart catheterization. With these limitations in mind, the investigation revealed the mean age of patients with pulmonary hypertension of any etiology was 68.5 years and 54.5% were female. The overall group included 13.8% with WHO Group 1, 68.5% with Group 2, 47% with Group 3 and 9% with Group 4 PH. Mixed WHO Group characteristics were suggested in 35.4% of patients who had diagnosis codes signifying more than one WHO Group. This study demonstrated a rise in both incidence and prevalence of PH during the decade from 2002 to 2012. Mortality rates for the overall PH population were 12.8%, 35.9% and 61.5% at 30 days, 1 year and 5 years, respectively. Patients with WHO Group 1 PH had the lowest mortality rate, while those with WHO Group 2 and 3 had the highest risk of death. Other reports also indicate that left heart disease is the most common cause of PH with COPD being the second most common worldwide.26
Population registries from France and the United States have illustrated the epidemiologic makeup of WHO Group 1 PAH.27,28 As noted previously, pulmonary arterial hypertension can be subclassified depending on whether it is heritable, idiopathic or associated with other conditions which are considered risk factors for PAH. Entities considered risk factors for development of PAH include drug and toxin exposure, connective tissue disease, HIV infection, portal hypertension, congenital heart disease and schistosomiasis. There is wide variability in reports of incidence, prevalence, mortality and other characteristics of PAH depending on geographic differences in target populations, economic determinants, differences in health-care systems and clinical practice.29–33 The source for data acquisition can be an important factor contributing to variability as well. A recent comprehensive review of the literature pertaining to WHO Group 1 PAH and WHO Group 4 chronic thromboembolic pulmonary hypertension (CTEPH) underscores this point.29 Based on publications reflecting data from national registries, clinical databases and claims/administrative databases patients with PAH had a mean age ranging from 43 to 76 years of which 55% to 81% were female. The incidence of PAH was 1.5 to 32 cases per million of the general population and prevalence ranged from 12.4 to 268 cases per million. Similarly, with CTEPH wide variability was reported with mean age at diagnosis 58 to 73 years and 37% to 70% female. The incidence of CTEPH from this review ranged from 0.9 to 39 cases per population million and prevalence ranged 14.5 to 144 per million.
Pathophysiology Behind Pulmonary Arterial Hypertension (WHO Group 1)
Patients classified with WHO Group 1 PAH share a vasculopathy of the small precapillary arteries characterized by excessive endothelial proliferation, smooth muscle proliferation and hypertrophy, in situ thrombosis, formation of vascular plexiform lesions.5,34 These vascular changes can also be found in the postcapillary vessels in certain disorders, such as systemic sclerosis and pulmonary veno-occlusive disease. In addition to characteristic remodeling of the pulmonary vasculature, there is a loss of precapillary arteries and exaggerated infiltration of the perivascular space with inflammatory cells.2 The pathogenesis of these changes is a complex process with evidence for genetic factors, cytokines and growth factors, ion channel dysfunction, and vascular injury with endothelial dysfunction resulting in imbalance of endogenous vasomotor regulation and cell proliferation in the pulmonary vascular bed.2,35,36
A genetic basis for PAH was first uncovered in 2000 with discovery of a bone morphogenetic protein receptor II (BMPR2) gene mutation in patients with heritable PAH.37,38 This mutation is identified in 70–80% of patients with heritable PAH and 15–25% with idiopathic PAH.39,40 BMPR2 is a member of the transforming growth factor-beta (TGF-B) gene superfamily and serves to limit smooth muscle cell proliferation and endothelial cell apoptosis.41 Sequence variations in genes encoding BMPR2-related downstream SMAD (Suppressor of Mothers Against Decapentaplegic) signaling intermediaries (SMAD1, SMAD4, SMAD8, SMAD9) have also been linked to the pathogenesis of PAH.42–44 Mutations involving activin receptor-like kinase 1 (ACVRL1 or ALK1) and endoglin (ENG) account for 80% of cases of hereditary hemorrhagic telangiectasia (HHT) with 15–20% of these patients also developing pulmonary hypertension.45,46 Other PAH-linked mutations unrelated to BMPR2 signaling involve KCNK3 and CAV1. KCNK3 (potassium channel subfamily K member 3) encodes a pH sensitive potassium channel that influences vascular tone.47 CAV1 (caveolin-1) encodes a membrane protein essential for creation of lipid rafts or caveoli that function to position BMP receptors.48 Up to 25% of sporadic cases of pulmonary capillary hemangiomatosis and pulmonary veno-occlusive disease have been attributed to mutations in EIF2AK4 (eukaryotic translation initiation factor 2 alpha kinase 4).49,50 In addition to mutations of the genome, epigenetic mechanisms affecting cellular function have also been a focus of recent research. These mechanisms may involve DNA methylation, micro RNAs or modification of histone proteins which alter expression of growth factor levels or gene expression and thereby influence cell growth and proliferation.51–54
Inflammatory cell infiltration is observed in proximity to areas of vascular remodeling in PAH raising interest in the potential role of cytokines and growth factors in the vascular disease process. It is unclear if the presence of inflammatory cell infiltrates represents a consequence of hypoxia, or alternatively if inflammatory mediators promote vascular cell injury and dysfunction. Examination of vascular lesions has demonstrated lymphocyte, macrophage, mast and dendritic cell invasion.55–57 While a deficiency of regulatory T cells has been noted in lungs of idiopathic PAH patients,58 an expansion of ectopic pulmonary lymphoid tissue and presence of autoantibodies suggest excessive B cell activation.59 Ectopic tissue adjacent to pulmonary arteries in PAH may be linked to the vascular remodeling process. Dendritic cells release cytokines that attract T and B lymphocytes, enhance their survival and contribute to an inflammatory environment.60 Macrophages located in the vicinity of vascular lesions are susceptible to the effects of IL-6 released by activated adventitial fibroblasts and assume a phenotype with proinflammatory characteristics.61 Macrophages are a source of platelet-derived growth factor (PDGF) which is a potent mitogen and chemoattractant for vascular cells.62 Although the exact role of immune cell biology and inflammation in the pathogenesis of PAH is yet to be defined, the presence of similar pulmonary vascular changes in both PAH and a significant number of patients with primary autoimmune disorders supports a role for immune cell mediated events in pulmonary vascular remodeling.
Aside from the impact of genetic and immune or inflammatory factors on structural remodeling, there is evidence that endothelial injury and dysfunction cause imbalances in the production of endogenous mediators of vascular tone, platelet aggregation and cellular proliferation. Immunohistochemical studies have demonstrated significant reductions in nitric oxide synthase and prostacyclin synthase levels in pulmonary vascular endothelium where these enzymes serve critical roles in the production of nitric oxide and prostacyclin, both of which have vasodilatory and antiproliferative effects on pulmonary vascular cells.63,64 While production of nitric oxide and prostacyclin by pulmonary vascular endothelium is reduced, the production of endothelin-1 by endothelial cells is increased in PAH.65 Endothelin-1 promotes opposing properties including vasoconstriction and cell proliferation. Endothelin-1, survivin and vascular endothelial growth factor (VEGF) have been isolated in vascular plexiform lesions and are believed to enhance endothelial and smooth muscle cell proliferation while inhibiting apoptosis.65–67 Thromboxane production by the pulmonary endothelium is also increased leading to vasoconstriction and in situ thrombus formation within the pulmonary arteries.68
Other factors with potential roles in the pathogenesis of PAH include serotonin, autoantibodies, dysfunctional voltage-gated potassium channels, and cancer-like patterns of cellular proliferation and apoptosis resistance. Serotonin may promote vasoconstriction and vascular remodeling by stimulating smooth muscle cell (SMC) and fibroblast proliferation.69–71 Anti-endothelial cell and anti-fibroblast antibody expression against specific target antigens have been observed in idiopathic and scleroderma-related PAH, although the role of these autoimmune effectors in pathogenesis of PAH is unclear.72,73 Smooth muscle cell proliferation may be stimulated by serotonin transporter activation of the platelet-derived growth factor-beta (PDGF-B) receptor.74 Smooth muscle intracellular calcium regulates not only contraction, but also proliferation and resistance to apoptosis. Downregulation and dysfunction of voltage-gated potassium channels affect membrane polarization and increase calcium influx, which in turn promotes smooth muscle cell contraction and enhances proliferation by driving cells into the cell cycle.39,75,76 A number of cancer-like cellular behaviours have been observed in pulmonary vascular endothelial cells, SMCs and fibroblasts including monoclonal expansion of endothelial cells in idiopathic PAH, presence of unstable short DNA microsatellite sequences in plexiform lesions, and a persistent hyperproliferative and apoptosis-resistant state when endothelial cells are removed from the in vivo environment.77–79 Pulmonary vascular endothelial cells, SMCs and fibroblasts are more dependent on glycolysis for energy production.80–82 Mitochondria shift from glucose oxidation to uncoupled aerobic glycolysis in a manner similar to cancer cells, thus enhancing creation of precursors for DNA synthesis and rapid cell proliferation.83 Vascular remodeling in PAH is distinguished from cancer by the fact that pulmonary vascular cells have not been shown to reproduce in clonal fashion without control.
Pathophysiology Behind Pulmonary Hypertension in Other WHO Groups
Although pulmonary hypertension in non-PAH WHO Groups is typically thought to arise via mechanisms specific to other disease processes and their effects on the pulmonary circulation, there is evidence for similarities in pathophysiology across WHO Groups that stem from endothelial damage or dysfunction like that seen in PAH.35 Pulmonary hypertension in left heart disease (PH-LHD) is considered a consequence of the retrograde transmission of elevated left atrial pressure to the pulmonary circulation. Elevated left atrial pressure may be a consequence of systolic or diastolic left ventricular dysfunction or valvular heart disease. In some WHO Group 2 patients, vascular remodeling with characteristics similar to PAH may develop in distal pulmonary arteries and venules and persist even after correction of the underlying cause of left atrial hypertension.84 As an example, patients with valvular heart disease have been shown to exhibit indicators of persistent pulmonary hypertension after valvular repair and removal of the source of elevated post-capillary pressures.85 Endothelial dysfunction resulting from prolonged retrograde increases in pulmonary vascular pressure is thought to mediate many of the observed alterations in vascular structure and function.89 Elevated plasma endothelin levels are reported in patients having PH-LHD and nitric oxide production is reduced in heart failure.87,90 The pulmonary vasculopathy in PH-LHD is known to involve activation of fibroblasts, proliferation of the vascular intima and media, perivascular infiltration with inflammatory cells, and increased cytokine and growth factor expression all similar to features seen in PAH.86–88 These findings correlate with observations of vascular remodeling resembling that seen in PAH.
WHO Group 3 pulmonary hypertension due to lung disease and hypoxia is often considered a result of hypoxic vasoconstriction and loss of small vessels and capillaries. While this is true, chronic hypoxia has also been linked to the activity of several mediators affecting vascular structure and function similar to the vascular remodeling noted in PAH.91,92 Supplemental oxygen may be helpful in reversing hypoxic vasoconstriction in early stages, but as disease progresses and hypoxia becomes a more chronic fixture, vascular remodeling becomes a more significant cause of rising pulmonary vascular resistance. As with PAH, endothelial biology plays a key role in the pulmonary vascular response to hypoxia and is influenced by TGF-B1 and hypoxia-inducible factor 1-alpha (HIF-1a) signaling pathways.81,91,93,94 In experimental models, chronic hypoxia induces expression of vascular endothelial growth factor A (VEGFA) and its receptor VEGFR2 via HIF-1a.91,95,96 Hypoxia also induces expression of TGF-B1 and thereby enhances production of PDGF-B. PDGF-B has been shown to promote VEGFA expression and hence hypoxia-induced endothelial proliferation.91,97 Chronic hypoxia is a stimulus for serotonin release, which in turn stimulates vasoconstriction and promotes endothelial proliferation and smooth muscle hypertrophy.92,98 Hypoxia is reported to decrease endothelial cell nitric oxide production and increase endothelin release.92,99 As a clinical correlate, impaired endothelial-dependent SMC relaxation has been observed in patients with COPD of varying severity.100 There is considerable interest in the relationship of tobacco smoking to vascular remodeling and pulmonary hypertension in chronic lung disease. Smoking by its own virtue appears to have multiple effects on lung vascular biology including enhanced VEGF expression, reduced nitric oxide synthase levels, increased infiltration of inflammatory cells, endothelial hyperplasia and disrupted mitochondrial function.101–103 Although poorly understood overall, it is clear that the root causes of pulmonary hypertension in the patient with chronic lung disease and hypoxia go much deeper than hypoxic vasoconstriction and loss of small vessels.
Pulmonary hypertension is a well-known complication of acute pulmonary thromboembolism. In the acute phase of thromboembolism pulmonary vascular resistance may be increased by significant clot burden obstructing the pulmonary vascular bed. Typically, with thrombolysis and/or anticoagulation, the obstructing clot will be eliminated and normal vascular resistance restored. However, even with continuous anticoagulation, lung perfusion defects can persist beyond 3 months from an acute event in over 50% of cases.104 A small percentage of these patients will go on to develop WHO Group 4 chronic thromboembolic pulmonary hypertension (CTEPH). Studies estimate the annual incidence of CTEPH after an acute pulmonary embolism to range from 0.4% to 6.2%.105 The true incidence is uncertain though, because current estimates of CTEPH incidence after acute PE may include patients that already had chronic thromboembolism prior to the acute event. CTEPH may be diagnosed several months or years after continuous anticoagulation and without symptomatic recurrent acute events. Pulmonary hypertension in this population is, in part, a consequence of nonresolving thrombus. In contrast to the characteristic fresh clot seen in acute pulmonary embolism, the chronic flow limiting material in CTEPH consists of a yellow fibrotic material that adheres tightly to the vessel wall and is composed of collagen, elastin, inflammatory cells and recanalization vessels.105
There are several proposed theories pertaining to the non-resolution of thrombotic material in patients who develop CTEPH. When the thromboembolic burden is large, the intrinsic lytic system may be unable to achieve complete resolution due to insufficient lytic capacity or ability to reach the entire mass of clot. Another possibility is that patients who are treated may not be anticoagulated adequately or long enough. Underlying autoimmune disorders, non-O blood group, history of splenectomy, ventriculo-atrial shunts, thyroid replacement and a history of malignancy were all associated with higher CTEPH risk in a large study comparing patients with CTEPH to those with other forms of pulmonary hypertension.106 Higher levels of several inflammatory markers and mediators of inflammation have been reported in CTEPH suggesting that underlying inflammation is somehow involved.107,108 Mutations causing abnormal fibrinogen and hence abnormal fibrin structure and resistance to plasmin-mediated lysis have been identified.109–111 Abnormalities of platelet function and dysfunctional angiogenesis and recanalization of thrombus have also been suggested to explain nonresolution of thrombus in CTEPH.112,113
Nonresolution of clot alone does not explain the development of pulmonary hypertension in CTEPH. Small vessel remodeling similar to that described in idiopathic PAH is also observed in CTEPH and involves vessels with unobstructed flow, as well as those distal to flow-limiting thrombi. Further, the characteristics of arterial vasculopathy extend to involve venules and small veins. The development of vasculopathy in unobstructed arteries has been considered a consequence of redirected flow with increased pressure and shear stress leading to endothelial injury.114 However, this mechanism would not explain development of an arteriopathy distal to obstructive thrombi. Anastamoses between the bronchial and pulmonary arteries have been identified that may allow flow distal to pulmonary arterial obstructions.115 It is theorized that exposing small arteries distal to thrombus to systemic level pressures may promote vascular remodeling in these arteries. Aside from these mechanical considerations, there is evidence that molecular factors favoring vascular remodeling also exist in CTEPH. Levels of the endogenous vasodilator, nitric oxide, are known to be reduced in both patients with PAH and CTEPH.116 Further, levels of the nitric oxide synthase inhibitor, asymmetric dimethylarginine, are increased and may limit the favorable vasorelaxant and antiproliferative actions of nitric oxide at the vascular level.117 Increased levels of endothelin, as noted PAH, have also been observed in CTEPH patients.118 Even though thrombosis is the primary event in CTEPH, there is growing evidence that development of pulmonary hypertension involves aberrations in a complex molecular signalling system similar in many ways to that seen in PAH.
Risk Assessment and Treatment of WHO Group 1 PAH
Treatment options for pulmonary arterial hypertension have rapidly evolved in recent years. Prior to 1995 treatment options for patients with PAH included diuretics, anticoagulation, digoxin, calcium channel blockers and oxygen. Those patients who had unfettered disease progression might ultimately require lung transplantation. In 1995 epoprostenol was the first targeted therapy approved by the United States Federal Drug Administration for treatment of PAH. Several additional targeted therapies have been approved since that time, expanding the number of non-surgical options available to patients. Currently available treatment agents for PAH target pathophysiologic mechanisms in nitric oxide, endothelin, or prostacyclin pathways. As more medical therapies have become available, so has evidence that combining agents and targeting multiple pathways simultaneously is often more beneficial than using them individually. Extending the use of WHO Group 1 PAH targeted therapies for treatment in other WHO Groups has met with limited success, leaving a paucity of good options for treating these patients. Therefore, most of the focus on available treatments in this review will pertain to options for WHO Group 1 PAH and Group 4 CTEPH with mention of recent investigations into extension of the agents for use in other WHO Groups.
Choosing from among the available medical therapies to devise an effective PAH treatment plan involves an ongoing process of risk assessment and outcome monitoring. PAH is a progressive disease, even in the current era of targeted therapy, and careful risk and outcome monitoring is an essential part of the treatment process. The observation that there is a strong correlation between survival and certain clinical characteristics in PAH, such as 6 minute walk distance, functional class, hemodynamic measures and other parameters, has led to the development of several risk assessment tools.119 Right ventricular function is widely recognized as a determinant of outcome in PAH. Noninvasive measures of right ventricular structure and function by echocardiography or cardiac MRI are useful in risk stratification and have been incorporated into management guidelines.120,121 The recent European Society of Cardiology/European Respiratory Society guidelines include right atrial area, tricuspid annular plane systolic excursion/systolic pulmonary artery pressure (TAPSE/sPAP) and presence of pericardial effusion among risk stratification variables.3 Risk assessment tools integrate multiple clinical parameters to predict whether a patient is at low, moderate or high risk of near-term death from PAH, and as such, guide the approach to medical treatment design. Two of the most commonly used risk assessment tools are the ESC/ERS risk assessment algorithm3 and the REVEAL risk calculator.122 The REVEAL risk calculator was created from data acquired in the American REVEAL PAH Registry which was a 3-year longitudinal registry of 2967 WHO Group 1 PAH patients with data collected pertaining to clinical characteristics, evaluation, treatment and outcomes.123 A validated revision of the REVEAL calculator (REVEAL 2.0) includes an abridged version (REVEAL Lite 2) that improves usefulness in outpatient settings were regular follow up care is provided.124,125 Risk assessment is typically performed at each regular follow up visit, and adjustments to the treatment plan are made as needed to achieve and maintain low risk status. Based on REVEAL 2.0 risk stratification, patients in the low-risk group have a > 94% predicted 1-year survival, moderate-risk 70 to <94% 1-year survival and high-risk <70% 1-year survival. Current treatment guidelines incorporate risk assessment in the algorithm to modify treatment plans.
Nitric Oxide Pathway
Nitric oxide (NO) is an endogenous vasodilator produced by the pulmonary endothelium. Nitric oxide synthase (NOS) converts L-arginine to NO which acts on adjacent smooth muscle cells where it catalyzes conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP) by guanylate cyclase. Cyclic GMP in turn promotes smooth muscle relaxation and vasodilation, but also regulates cell proliferation, apoptosis and inflammation.126,127 Cyclic GMP is neutralized by endogenous phosphodiesterase-5 (PDE-5), thus limiting its effects. Patients with PAH have deficient NOS activity in the pulmonary vasculature leading to low levels of NO production.63 Favorable outcomes in the treatment of PAH have been achieved by targeting the NO-cGMP pathway with agents that inhibit PDE-5 (PDE-5 inhibitors) or directly stimulate guanylate cyclase (soluble guanylate cyclase stimulators).
Phosphodiesterase-5 inhibitors limit the inactivation of cGMP, thereby augmenting its impact in vascular smooth muscle. Three PDE-5 inhibitors, sildenafil, tadalafil and vardenafil, have been shown to improve pulmonary hemodynamics, exercise capacity and symptoms in patients with PAH, including those with connective tissue disease.128–130 Additionally, tadalafil has been shown to increase the time to clinical worsening.129 These agents may precipitate severe hypotension if used in conjunction with nitroglycerin, and therefore the use of nitroglycerin with PDE-5 inhibitors is contraindicated. Safety in pregnant humans has not been established; however, fetal harm was not noted in animal studies.
Guanylate cyclase can be directly stimulated to produce cGMP by soluble guanylate cyclase (sGC) stimulators. Riociguat is the only sGC stimulator approved for the treatment of pulmonary hypertension to date, and has been approved by the US Federal Drug Administration for both PAH and CTEPH. The benefits of riociguat for pulmonary hypertension have been elucidated in two randomized clinical trials. In PATENT-1, riociguat improved hemodynamics, exercise capacity and time to clinical worsening in patients with PAH.131 In CHEST-1 riociguat was associated with significant improvement in exercise capacity and pulmonary vascular resistance in patients with CTEPH.132 Riociguat is currently the only therapeutic agent approved for treatment of CTEPH (WHO Group 4 PH). Like the PDE-5 inhibitors, riociguat can cause hypotension if used with nitroglycerin. Thus, using riociguat and nitroglycerin together is contraindicated. Further, riociguat is teratogenic and should not be used in pregnant humans. Females of childbearing age are required to have monthly pregnancy testing and follow careful contraceptive measures while taking riociguat.
Endothelin Pathway
Endothelin-1 is a vasoconstrictor and mitogenic mediator produced by endothelial cells and present at increased levels in plasma and lung tissue of patients with PAH.65,133 Endothelin exerts its effects by binding to two G protein-coupled receptors, types A and B, which are located on the smooth muscle cell surface. The type A receptor mediates vasoconstriction, cell growth and inflammation, while the B type receptor promotes vasodilation and natriuresis while limiting cell proliferation and inflammation. There are three compounds in this group used for the treatment of PAH, including bosentan, ambrisentan, and macitentan.
Bosentan blocks both type A and B receptors to improve exercise capacity, time to clinical worsening, hemodynamics and echocardiographic abnormalities in PAH.134 Bosentan use was associated with liver transaminase elevations in about 10% of patients in clinical trials leading to a requirement for liver function monitoring on a monthly basis. In addition, bosentan exposure has been associated with edema and anemia. The drug is teratogenic and should not be used in pregnant humans. Females of reproductive age are required to undergo monthly pregnancy testing and practice careful contraception.
In contrast to bosentan, ambrisentan is a selective type A receptor antagonist which was associated with significant improvements in symptoms, exercise capacity, clinical worsening and hemodynamics in clinical trials including patients with idiopathic PAH or PAH related to connective tissue disease or HIV infection.135,136 The risk of liver injury is minimal with ambrisentan, and monthly liver function testing is not required. However, using ambrisentan in patients with chronic liver disease should be avoided. Again, ambrisentan is teratogenic leading to the same pregnancy and contraceptive precautions as with bosentan.
The third, and newest, endothelin receptor antagonist is macitentan. The benefits of macitentan in patients with PAH was demonstrated in a large event-driven trial during which macitentan reduced the risk of disease progression by 45% and the risk of death or hospitalization due to PAH by 50%.137 A significant number of patients were on background therapy with other agents in this study and, even so, demonstrated significant benefit from macitentan. The major adverse effects of this drug include anemia and fluid retention. Monitoring for liver injury is not required, however like other endothelin receptor antagonists, macitentan is teratogenic and should not be used in pregnant females. Pregnancy monitoring and careful contraceptive measures are required in females with reproductive capacity.
Prostacyclin Pathway
Prostaglandin I2 produced from arachidonic acid in the pulmonary endothelial cell mediates smooth muscle cell relaxation by catalyzing the production of cyclic adenosine monophosphate (cAMP) from adenosine triphosphate (ATP) in smooth muscle cells. Cyclic AMP mediates smooth muscle relaxation, vasodilation and antiproliferative activities in the pulmonary vascular bed. The prostacyclin analogues are potent vasodilators of the pulmonary vasculature, inhibitors of platelet aggregation and inhibitors of cell proliferation that exert their effect by interaction with prostaglandin receptors.138 Pulmonary endothelial prostacyclin synthase expression is reduced in PAH resulting in reduced production of prostaglandin I2 from arachidonic acid and thereby reduced smooth muscle cyclic AMP production.64 Synthetic prostacyclin analogues can offset the deficiency of endogenous prostaglandin I2 production in PAH and are available as oral, inhaled, and subcutaneously or intravenously delivered disease-modifying therapies.
Epoprostenol was the first prostacyclin analogue shown to be effective in reducing symptoms, improving exercise capacity, and improving hemodynamics in idiopathic PAH and PAH associated with systemic sclerosis.139,140 Further, treatment with epoprostenol was noted to reduce mortality in one randomized trial involving idiopathic PAH patients.140 Sustained long-term benefits of epoprostenol have been demonstrated in idiopathic PAH, PAH associated with other disorders and inoperable CTEPH.141–145 Epoprostenol is only available as a continuous IV therapy and has a short half-life of 3–5 minutes. Tachyphylaxis occurs with the continuous infusion requiring dose escalation over time. The infusion is typically well tolerated, although adverse effects including jaw pain, headache, flushing, nausea and diarrhea may be experienced for a short period after dose escalation. There is a serious risk of rebound vasoconstriction and even death if the infusion is disrupted.
Treprostinil is a newer prostacyclin analogue that is available in oral, inhaled, and subcutaneously or intravenously infused forms. Treprostinil was first investigated using a subcutaneous delivery method in a large group of patients with idiopathic PAH and PAH associated with connective tissue disease. Although dose titration was limited by adverse effects, such as infusion site pain, the study demonstrated improvement in exercise tolerance and hemodynamics.146,147 The use of treprostinil for PAH was later extended to include an intravenous application. The infused forms of treprostinil are associated with tachyphylaxis, similar to epoprostenol, requiring ongoing monitoring and dose escalation. The half-life of treprostinil is 3–4 hours. Adverse effects are similar to those noted with epoprostenol and include the added risk of infusion site pain for those using the subcutaneous route of treatment. The inhaled formulation of treprostinil is an effective treatment option for those patients who may not be suited for a continuous infusion system. Significant improvements in 6 minute walk distance, NT-proBNP levels and quality of life indicators were observed in a randomized clinical trial of inhaled treprostinil that included patients already on background therapy with bosentan or sildenafil.148 Inhaled treprostinil is administered four times daily and, as such, is not associated with development of tachyphylaxis. Inhaled treprostinil is well tolerated with the most common adverse effects including sore throat, cough and headache. An oral formulation of treprostinil has also been shown in a randomized clinical trial of treatment naïve PAH patients to improve 6 minute walk distance.149 Oral treprostinil is delivered on a b.i.d. or t.i.d. schedule with gradual dose escalation to achieve and maintain optimal effect. Use of oral treprostinil may be complicated by anorexia, nausea and diarrhea. The oral formulation should not be used in patients with Child Pugh Class 3 hepatic impairment.
Iloprost is a prostacyclin analogue available in Europe in intravenous and inhaled formulations but only available in the inhaled form in the United States. The inhaled formulation was compared with placebo in patients with PAH and CTEPH and led to improved symptoms, exercise capacity and hemodynamics.150 Benefits of intravenous iloprost were found to be similar to those of epoprostenol in a small group of patients with either PAH or CTEPH.151 Inhaled iloprost is delivered with a specifically designed nebulizer and requires treatment 6 to 9 times a day. Each dose is effective for 30–90 minutes and may be associated with cough, flushing or jaw pain. Hypotension may occur, so caution should be exercised in patients with lower baseline blood pressure.
In addition to exogenous delivery of prostacyclin analogues, the unfavorable effect of prostacyclin synthase deficiency in PAH can also be overcome with a newer targeted therapy class, the prostacyclin receptor agonist. Prostanoid receptors within the pulmonary artery include IP, EP3 and TP receptors. The IP receptor mediates vasodilation and limits proliferation, while the others promote vasoconstriction and cell proliferation.152–154 To date, selexipag, is the only commercially available prostacyclin receptor agonist, although a second agent, ralinepag, is currently under investigation in patients with PAH.
Selexipag is metabolized to an active form that is 37 times more potent than the parent compound. In a long-term event-driven investigation, selexipag reduced the risk of a composite morbidity and mortality endpoint by 40%.155,156 A similar composite endpoint risk reduction of 41% was noted in a subgroup of patients with PAH related to connective tissue disease.157 Composite endpoint events included need for atrial septostomy or lung transplantation, initiation of chronic oxygen or parenteral prostanoid therapy, hospitalization for a PAH related reason, other indicator of disease progression, or death. A large majority (80%) of patients were on background therapy with an endothelin receptor blocker, PDE- inhibitor or both. A consistent treatment effect was noted irrespective of background therapy. Common adverse effects of this particular agent include headache, flushing, nausea, diarrhea, myalgia, and arthralgia.
Combination Therapy
While the various therapeutic agents used to treat PAH are effective in their own right, data has accumulated indicating that combinations of agents from different classes may be even more effective in limiting disease progression. There have been numerous clinical trials in recent years that have examined the benefits of combination therapy and demons...



Comments
Post a Comment